
The FDA has approved the first blood test to detect signs of Alzheimer’s disease, marking a major step toward easier diagnosis.
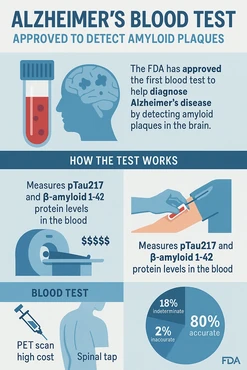
The Lumipulse G pTau217/ß-Amyloid 1-42 Plasma Ratio test identifies amyloid plaques through a simple blood draw, reducing reliance on expensive PET scans and invasive spinal taps.
Alzheimer's impacts nearly 7 million Americans, with projections hitting 13 million by 2050 — early, accessible detection could be game-changing.
The U.S. Food and Drug Administration (FDA) today cleared the first in vitro diagnostic device that tests blood to aid in diagnosing Alzheimer’s disease. The test — officially called the Lumipulse G pTau217/ß-Amyloid 1-42 Plasma Ratio — is for the early detection of amyloid plaques associated with Alzheimer’s disease in adult patients, aged 55 years and older, exhibiting signs and symptoms of the disease.
“Alzheimer’s disease impacts too many people, more than breast cancer and prostate cancer combined,” said FDA Commissioner Martin A. Makary, M.D., M.P.H. “Knowing that 10% of people aged 65 and older have Alzheimer's, and that by 2050 that number is expected to double, I am hopeful that new medical products such as this one will help patients.”
Alzheimer’s disease, a brain disorder known to slowly destroy memory and thinking skills, and, eventually, the ability to carry out the simplest tasks, is progressive, meaning that the disease gets worse over time. In most people with Alzheimer’s disease, clinical symptoms first appear later in life.
Amyloid plaques in a patient’s brain are a hallmark sign of Alzheimer’s disease. While amyloid plaques can occur in other diseases, being able to detect the presence of plaque, along with other evaluations, helps the doctor determine the probable cause of the patient’s symptoms and findings. These plaques can be detected early using positron emission tomography (PET) brain scans but PET scans are costly and time-consuming and expose patients to radiation.
Measures two proteins
The Lumipulse/Amyloid ratio measures two proteins found in human plasma and calculates the numerical ratio of the levels of the two proteins. This ratio is correlated to the presence or absence of amyloid plaques in the patient’s brain, reducing the need for a PET scan. Similar tests are used with cerebrospinal fluid (CSF) samples, which are collected through an invasive lumbar puncture, also called a spinal tap. This new Lumipulse test only requires a simple blood draw, making it less invasive and much easier for patients to access.
“Nearly 7 million Americans are living with Alzheimer's disease and this number is projected to rise to nearly 13 million,” said Center for Devices and Radiological Health Director Michelle Tarver, M.D., Ph.D. “Today’s clearance is an important step for Alzheimer’s disease diagnosis, making it easier and potentially more accessible for U.S. patients earlier in the disease.”
About the review process
During review of the new test, the FDA evaluated data from a multi-center clinical study of 499 individual plasma samples from adults who were cognitively impaired. The samples were tested by the Lumipulse/Amyloid ratio and compared with amyloid PET scan or CSF test results.
In this clinical study, 91.7% of individuals with positive results on the Lumipulse/Amyloid ratio had the presence of amyloid plaques by PET scan or CSF test result, and 97.3 % of individuals with negative results had a negative amyloid PET scan or CSF test result. Less than 20% of the 499 patients received an indeterminate result.
These findings indicate that the new blood test can reliably predict the presence or absence of amyloid pathology associated with Alzheimer’s disease at the time of the test in patients who are cognitively impaired. The test is intended for patients presenting at a specialized care setting with signs and symptoms of cognitive decline.
The FDA issued clearance of the Lumipulse G pTau217/ß-Amyloid 1-42 Plasma Ratio to Fujirebio Diagnostics, Inc.