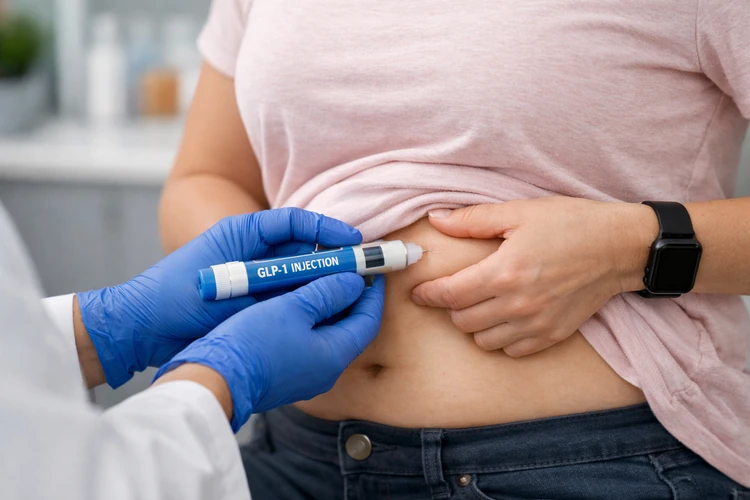
Zepbound is Eli Lilly’s weight loss drug. Now, it is also a drug to treat obstructive sleep apnea (OSA).
The U.S. Food and Drug Administration has approved Zepbound (tirzepatide) as the first drug treatment for adults with moderate to severe OSA who also have obesity. The FDA said the approval marks a significant advancement in the management of a condition that affects millions of individuals, particularly those struggling with obesity.
Dr. Sally Seymour, director of the Division of Pulmonology, Allergy, and Critical Care in the FDA’s Center for Drug Evaluation and Research, said the approval “is a major step forward for patients with obstructive sleep apnea.”
Obstructive sleep apnea is characterized by repeated interruptions in breathing during sleep due to the blockage of the upper airway. While continuous positive airway pressure (CPAP) devices have been the standard treatment, many patients find them uncomfortable or difficult to use consistently.
A new approach to an old problem
The FDA said Zepbound offers a new pharmacological approach by activating receptors of hormones secreted from the intestine, specifically glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), to reduce appetite and food intake, thereby promoting weight loss and improving OSA symptoms.
The FDA said its approval is based on the results of two pivotal randomized, double-blind, placebo-controlled studies involving 469 adults without type 2 diabetes. These studies demonstrated that participants receiving Zepbound experienced a significant reduction in the apnea-hypopnea index (AHI), a key measure of OSA severity, compared to those receiving a placebo.
The studies also showed that a greater proportion of participants treated with Zepbound achieved remission or mild OSA, alongside significant weight loss.
Potential side effects
The FDA cautions that Zepbound is not without potential side effects. Common adverse reactions include gastrointestinal issues such as nausea, diarrhea, and abdominal discomfort, as well as injection site reactions and fatigue.
More serious concerns include the risk of thyroid C-cell tumors, pancreatitis, gallbladder problems, and hypoglycemia, particularly when used with insulin or insulin-secreting medications. Consequently, Zepbound is contraindicated in patients with a personal or family history of medullary thyroid cancer or Multiple Endocrine Neoplasia syndrome type 2.
The FDA said patients considering Zepbound should engage in thorough discussions with their healthcare providers to weigh the benefits against potential risks, especially if they have pre-existing conditions such as kidney disease, diabetic retinopathy, or a history of depression or suicidal thoughts.